In medicine, the CDAGS syndrome is a rare complex of malformations that is primarily associated with cranial anomalies. The symptom complex has a hereditary basis and is caused by mutation on chromosome 22. A causal therapy does not yet exist.
What is CDAGS Syndrome?
CDAGS syndrome is also known as CAP syndrome and is an extremely rare hereditary disease. The prevalence is estimated to be one in 1,000,000 people. The disease was first described in the 21st century. R. Mendoza-Londono and his colleagues are considered to be the first to describe it and give it its name. The term CDAGS syndrome is an acronym.
This acronym contains the clinical combination of features that characterize the symptom complex. The C stands in this connection for craniosynostosis, which shortens D “delayed” and refers to a delayed closure of the fontanelle or the “deafness”, ie numbness of patients. A codes the anal malformations associated with the syndrome.
The G is given the acronym due to the genital malformation with which the disease is symptomatically associated and the S stands for “skin” and indicates the dermal changes within the clinical picture. The syndrome falls into the group of unspecified malformation syndromes and corresponds to a congenital disease.
Causes
The CDAGS syndrome has a genetic basis. The syndrome does not seem to occur sporadically, but rather to be inherited in an autosomal recessive manner. So far, the complex of symptoms has only been described within four families. In all of them there was a familial accumulation, which confirms the assumption of a genetic component.
The symptoms were found in a total of seven individuals within the four families. A hereditary mutation is the cause of the symptom complex. Despite the few documented cases so far, the genetic cause has now been linked to certain genes. The mutation affects the genes on chromosome 22 at the gene locus 22q12-q13.
Chromosome 22 is the second smallest chromosome in humans and contains between 500 and 800 genes, of which 508 are known to date. Mutations in the genes on chromosome 22 are implicated in many different diseases. Examples are trisomy 22, Epstein syndrome and Sebastian syndrome.
Which genes of the chromosome can play a role in the development of the CDAGS syndrome is the subject of research. At present one can only speculate about the primary causal factors of the syndrome. In addition to poison exposure during pregnancy, numerous other factors can be considered as activating factors for the genetic disposition.
Symptoms, ailments & signs
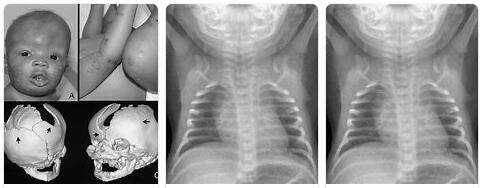
Cranial abnormalities shape the clinical picture of CDAGS syndrome. These are deformities that affect the skull. The synostosis of the coronary suture is particularly characteristic of the syndrome. The term synostosis refers to a bony fusion of several bones that were formerly cartilaginous or connected by connective tissue.
The synostosis of the cranial suture in patients with CDAGS syndrome is associated with wide-open fontanelles in the anterior and posterior area. The parietal foramina of the patient is also noticeably large. The parietal foramen belongs to the parietal bone and corresponds to an opening in the upper parietal bone through which the parietal emissary vein passes. In addition to these anomalies of the skull, those affected usually suffer from keratinization and thus cornification disorders in the form of porokeratosis, which appears as a dermal rash.
Often those affected are also affected by sensorineural hearing loss or even absolute deafness. Anal and urogenital anomalies complete the picture. These symptoms are associated with severe delays in intellectual and motor development. Under certain circumstances, there is also an underdevelopment of the collarbones.
Diagnosis
CDAGS syndrome is usually diagnosed in infancy. The first suspicion overtakes the doctor through eye diagnosis. Imaging of the skull may be ordered to confirm the suspicion, which will provide evidence of the cranial abnormalities.
Dermatological tests can also take place as part of the diagnosis. To make the diagnosis unequivocal, the doctor can order molecular genetic tests. A mutation on chromosome 22 confirms the suspicion of the syndrome.
Complications
Due to the CDAGS syndrome, the patient is affected by various malformations and deformities. In this case, these have a negative effect on the skull in particular and can lead to discomfort in the bones. The head is noticeably large, which leads to reduced aesthetics. In many cases, teasing and bullying occur, especially among children.
This can lead to psychological complaints. It is not uncommon for CDAGS syndrome to have reduced hearing or even complete deafness. This can also put a heavy strain on the person’s life and everyday life. However, there is no reduction in intelligence, so that the person concerned usually understands and can understand thought processes. A causal treatment of CDAGS syndrome is not possible.
Only the symptoms can be limited, but there are no further complications. The symptoms are limited by surgical interventions or with the help of various therapies. However, it cannot be predicted whether these will lead to a positive course of the disease. CDAGS syndrome does not reduce life expectancy in most cases.
When should you go to the doctor?
CDAGS syndrome is usually diagnosed in infancy. A doctor’s visit is necessary if the malformations and deformities cause complications. For example, if the child has bone pain or decreased hearing, the appropriate specialist must be contacted. The same applies to all minor and major complications associated with CDAGS syndrome. These must be treated in any case in order to enable the child to lead a symptom-free life despite the illness. Surgery may also be possible in the case of severe deformities.
Such an intervention must take place at an early stage, otherwise permanent developmental disorders can develop. If there is a suspicion of mental illness, triggered for example by teasing and bullying, the child should seek therapeutic advice together with the parents.
The general rule is: children and adults with CDAGS syndrome must be closely looked after by an experienced specialist. Since hereditary diseases can trigger unspecific symptoms, various medical professionals should be consulted who can give the person affected a good picture of the symptoms and possible therapeutic methods.
Treatment & Therapy
So far, no causal treatment measures are available to patients with CDAGS syndrome. For one thing, the chromosome has been identified, but not the responsible gene. On the other hand, gene therapy has so far been a subject of research, but is not yet applicable. For this reason, patients with the syndrome are treated symptomatically.
This symptomatic treatment includes, for example, corrections of the cranial anomalies, which are usually carried out surgically. The synostosis can only be surgically resolved if there are clearly problems that can be traced back to the adhesions. The intervention gives the brain more space to grow. If the surgery is done early enough, it can alleviate the developmental disorders of the patient.
The anal and urogenital anomalies in the patient can usually be improved by surgery. However, these steps are usually less urgent than the skull correction, since restricted growth of the brain is associated with far more secondary symptoms.
The porokeratosis of those affected can be treated using conventional medication, for example by administering keratolytics that loosen the horny layer. To compensate for mental and motor development delays, early intervention and physiotherapy can be useful.
Outlook & forecast
CDAGS syndrome cannot be treated with causal therapy. For this reason, only symptomatic treatment options are available to the person concerned, but these cannot completely limit the disease.
The various malformations and malformations can be corrected with the help of surgical interventions. If there is no treatment, these malformations and adhesions remain and lead to severe restrictions in the life of the person concerned. Early diagnosis and treatment of CDAGS syndrome can reduce and minimize disruptions in the development of those affected.
If the skull correction does not take place, the brain can usually not grow easily, so that it can lead to mental disorders and in the worst case to brain death. With timely treatment, these complaints can be prevented.
The other symptoms are usually alleviated with the help of medication. Since the CDAGS syndrome also leads to reduced intelligence and to motor and mental complaints, these are compensated for by special support. However, a complete healing cannot take place here either. However, with early therapy, CDAGS syndrome does not have a negative impact on the patient’s life expectancy.
Prevention
The CDAGS syndrome cannot yet be prevented. For example, the primary causal factors are not currently known in detail, so that the scope for action is limited.
You can do that yourself
Before self-help measures are taken, those affected by CDGAS syndrome should first speak to the responsible doctor. Usually the individual symptoms and ailments can only be treated surgically, and personal measures such as applying ointments to the abscesses often cause secondary symptoms. It is therefore important to clarify the next steps with a specialist.
The doctor will primarily recommend a healthy lifestyle and regular exercise. In addition, physiotherapeutic measures are indicated. Physiotherapy, yoga and the like help to stabilize the immune system and avoid long-term effects such as hemorrhoids on the anus or abscesses on the skull. Depending on the severity of the CDGA syndrome, it may also be necessary to promote mental abilities. This should always be done under professional supervision. However, the relatives and friends of the person concerned can support the respective therapy – in which way depends on the individual complaints and the constitution of the patient.
Spiritual support is also important. Since those affected usually suffer very much from the anomalies, emotional support is all the more important, especially in the case of mental limitations or a poor general prognosis.